Node.js provides a simple mechanism to write a REST server. As an exercise, I wrote a REST server for the mysql server of the UCSC genome bowser. The code is available on github at:
Starting the server
$ cd bionode
$ node ucsc/ucsc.js
Server running at http://localhost:8080/
METHOD: /schema/databases
Lists the available databases :e.g: http://localhost:8080/schemas/databases
[
"information_schema",
"ailMel1",
"allMis1",
"anoCar1",
(...)
"visiGene",
"xenTro1",
"xenTro2",
"xenTro3"
]
This method accepts a parameter callback for JSON-P : e.g: http://localhost:8080/schemas/databases?callback=handle
handle([
"information_schema",
"ailMel1",
"allMis1",
"anoCar1",
(...)
"visiGene",
"xenTro1",
"xenTro2",
"xenTro3"
]);
METHOD: /schema/:database/tables
Lists the available tables for a given database :e.g: http://localhost:8080/schemas/anoCar1/tables
[
"all_mrna",
"author",
"blastHg18KG",
"cds",
(...)
"xenoRefFlat",
"xenoRefGene",
"xenoRefSeqAli"
]
This method accepts a parameter callback for JSON-P : e.g: http://localhost:8080/schemas/anoCar1/tables?callback=handle
handle([
"all_mrna",
"author",
"blastHg18KG",
"cds",
"cell",
(...)
"xenoRefFlat",
"xenoRefGene",
"xenoRefSeqAli"
]);
METHOD: /schema/:database/:table
Returns a schema for the given database.table. E.g: http://localhost:8080/schemas/anoCar1/xenoMrna
{"database":"anoCar1","table":"xenoMrna","fields":[{"name":"bin","type":"smallint(5) unsigned","key":""},{"name":"matches","type":"int(10) unsigned","key":""},{"name":"misMatches","type":"int(10) unsigned","key":""},{"name":"repMatches","type":"int(10) unsigned","key":""},{"name":"nCount","type":"int(10) unsigned","key":""},{"name":"qNumInsert","type":"int(10) unsigned","key":""},{"name":"qBaseInsert","type":"int(10) unsigned","key":""},{"name":"tNumInsert","type":"int(10) unsigned","key":""},{"name":"tBaseInsert","type":"int(10) unsigned","key":""},{"name":"strand","type":"char(2)","key":""},{"name":"qName","type":"varchar(255)","key":"MUL"},{"name":"qSize","type":"int(10) unsigned","key":""},{"name":"qStart","type":"int(10) unsigned","key":""},{"name":"qEnd","type":"int(10) unsigned","key":""},{"name":"tName","type":"varchar(255)","key":"MUL"},{"name":"tSize","type":"int(10) unsigned","key":""},{"name":"tStart","type":"int(10) unsigned","key":""},{"name":"tEnd","type":"int(10) unsigned","key":""},{"name":"blockCount","type":"int(10) unsigned","key":""},{"name":"blockSizes","type":"longblob","key":""},{"name":"qStarts","type":"longblob","key":""},{"name":"tStarts","type":"longblob","key":""}]}
This method accepts a parameter callback for JSON-P : e.g: http://localhost:8080/schemas/anoCar1/xenoMrna?callback=handler
handler({"database":"anoCar1","table":"xenoMrna","fields":[{"name":"bin","type":"smallint(5) unsigned","key":""},{"name":"matches","type":"int(10) unsigned","key":""},{"name":"misMatches","type":"int(10) unsigned","key":""},{"name":"repMatches","type":"int(10) unsigned","key":""},{"name":"nCount","type":"int(10) unsigned","key":""},{"name":"qNumInsert","type":"int(10) unsigned","key":""},{"name":"qBaseInsert","type":"int(10) unsigned","key":""},{"name":"tNumInsert","type":"int(10) unsigned","key":""},{"name":"tBaseInsert","type":"int(10) unsigned","key":""},{"name":"strand","type":"char(2)","key":""},{"name":"qName","type":"varchar(255)","key":"MUL"},{"name":"qSize","type":"int(10) unsigned","key":""},{"name":"qStart","type":"int(10) unsigned","key":""},{"name":"qEnd","type":"int(10) unsigned","key":""},{"name":"tName","type":"varchar(255)","key":"MUL"},{"name":"tSize","type":"int(10) unsigned","key":""},{"name":"tStart","type":"int(10) unsigned","key":""},{"name":"tEnd","type":"int(10) unsigned","key":""},{"name":"blockCount","type":"int(10) unsigned","key":""},{"name":"blockSizes","type":"longblob","key":""},{"name":"qStarts","type":"longblob","key":""},{"name":"tStarts","type":"longblob","key":""}]});
METHOD: /ucsc/:database/:table/:column/:key
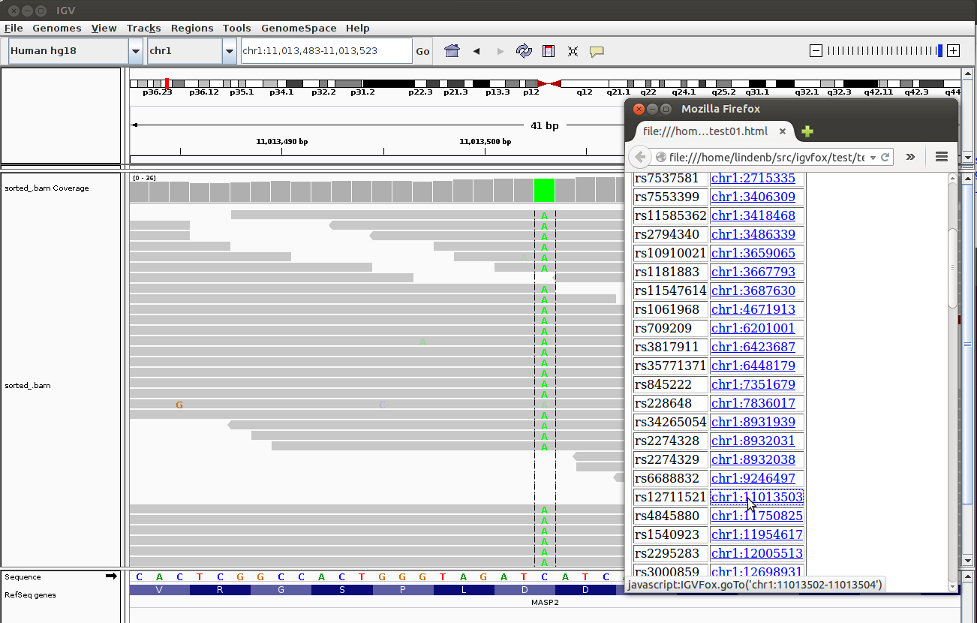
Fetch the rows for a given database.name having a :column==:key . The :column must be indexed. E.g: http://localhost:8080/ucsc/anoCar1/ensGene/name/ENSACAT00000004346
[
{"bin":592,"name":"ENSACAT00000004346","chrom":"scaffold_111","strand":"-","txStart":991522,"txEnd":996396,"cdsStart":991522,"cdsEnd":996396,"exonCount":3,"exonStarts":"991522,995669,995976,","exonEnds":"991954,995972,996396,","score":0,"name2":"PELO","cdsStartStat":"cmpl","cdsEndStat":"cmpl","exonFrames":"0,0,0,"}
]
This method accepts a parameter callback for JSON-P : e.g: http://localhost:8080/ucsc/anoCar1/ensGene/name/ENSACAT00000004346?callback=handler
handler([
{"bin":592,"name":"ENSACAT00000004346","chrom":"scaffold_111","strand":"-","txStart":991522,"txEnd":996396,"cdsStart":991522,"cdsEnd":996396,"exonCount":3,"exonStarts":"991522,995669,995976,","exonEnds":"991954,995972,996396,","score":0,"name2":"PELO","cdsStartStat":"cmpl","cdsEndStat":"cmpl","exonFrames":"0,0,0,"}
]);
METHOD: /ucsc/:database/:table?chrom=?&start=?&end=?
Fetch the rows for a given genomic database.name overlapping the given range. This method uses the UCSC-bin index if it is available; E.g: http://localhost:8080/ucsc/anoCar1/ensGene?chrom=scaffold_111&start=600000&end=900000
[
{"bin":589,"name":"ENSACAT00000003906","chrom":"scaffold_111","strand":"-","txStart":594783,"txEnd":614216,"cdsStart":595000,"cdsEnd":614201,"exonCount":9,"exonStarts":"594783,601291,601744,603640,604745,604865,609139,611740,614097,","exonEnds":"595105,601406,601813,603736,604771,604942,609173,611840,614216,","score":0,"name2":"DPM1","cdsStartStat":"cmpl","cdsEndStat":"cmpl","exonFrames":"0,2,2,2,0,1,0,2,0,"},
{"bin":589,"name":"ENSACAT00000003908","chrom":"scaffold_111","strand":"+","txStart":614382,"txEnd":615600,"cdsStart":614382,"cdsEnd":615600,"exonCount":1,"exonStarts":"614382,","exonEnds":"615600,","score":0,"name2":"MOCS3","cdsStartStat":"incmpl","cdsEndStat":"cmpl","exonFrames":"0,"},
{"bin":589,"name":"ENSACAT00000003918","chrom":"scaffold_111","strand":"-","txStart":638920,"txEnd":642127,"cdsStart":638920,"cdsEnd":642127,"exonCount":2,"exonStarts":"638920,641368,","exonEnds":"639691,642127,","score":0,"name2":"KCNG1","cdsStartStat":"cmpl","cdsEndStat":"cmpl","exonFrames":"0,0,"},
{"bin":591,"name":"ENSACAT00000003920","chrom":"scaffold_111","strand":"+","txStart":814576,"txEnd":826972,"cdsStart":814576,"cdsEnd":826972,"exonCount":3,"exonStarts":"814576,825125,826845,","exonEnds":"814594,825247,826972,","score":0,"name2":"ENSACAG00000003945","cdsStartStat":"incmpl","cdsEndStat":"cmpl","exonFrames":"0,0,2,"},
{"bin":591,"name":"ENSACAT00000004042","chrom":"scaffold_111","strand":"-","txStart":849731,"txEnd":881887,"cdsStart":849731,"cdsEnd":881887,"exonCount":24,"exonStarts":"849731,851343,855421,856165,857842,858090,861054,861943,862949,863773,865029,865639,867414,868216,872220,873601,874396,876850,877105,877711,878919,879681,881320,881738,","exonEnds":"849809,851460,855511,856279,857947,858201,861157,862027,863026,863866,865171,865722,867525,868368,872360,873738,874600,876994,877263,877850,878993,879847,881471,881887,","score":0,"name2":"ITGA2","cdsStartStat":"incmpl","cdsEndStat":"incmpl","exonFrames":"0,0,0,0,0,0,2,2,0,0,2,0,0,1,2,0,0,0,1,0,1,0,2,0,"},
{"bin":591,"name":"ENSACAT00000004050","chrom":"scaffold_111","strand":"-","txStart":883724,"txEnd":897808,"cdsStart":883724,"cdsEnd":897808,"exonCount":5,"exonStarts":"883724,885433,889264,889742,897701,","exonEnds":"883858,885548,889356,889852,897808,","score":0,"name2":"ENSACAG00000004086","cdsStartStat":"incmpl","cdsEndStat":"incmpl","exonFrames":"1,0,1,2,0,"}
]
This method accepts a parameter callback for JSON-P : e.g: http://localhost:8080/ucsc/anoCar1/ensGene?chrom=scaffold_111&start=600000&end=900000&callback=handler
handler([
{"bin":589,"name":"ENSACAT00000003906","chrom":"scaffold_111","strand":"-","txStart":594783,"txEnd":614216,"cdsStart":595000,"cdsEnd":614201,"exonCount":9,"exonStarts":"594783,601291,601744,603640,604745,604865,609139,611740,614097,","exonEnds":"595105,601406,601813,603736,604771,604942,609173,611840,614216,","score":0,"name2":"DPM1","cdsStartStat":"cmpl","cdsEndStat":"cmpl","exonFrames":"0,2,2,2,0,1,0,2,0,"},
{"bin":589,"name":"ENSACAT00000003908","chrom":"scaffold_111","strand":"+","txStart":614382,"txEnd":615600,"cdsStart":614382,"cdsEnd":615600,"exonCount":1,"exonStarts":"614382,","exonEnds":"615600,","score":0,"name2":"MOCS3","cdsStartStat":"incmpl","cdsEndStat":"cmpl","exonFrames":"0,"},
{"bin":589,"name":"ENSACAT00000003918","chrom":"scaffold_111","strand":"-","txStart":638920,"txEnd":642127,"cdsStart":638920,"cdsEnd":642127,"exonCount":2,"exonStarts":"638920,641368,","exonEnds":"639691,642127,","score":0,"name2":"KCNG1","cdsStartStat":"cmpl","cdsEndStat":"cmpl","exonFrames":"0,0,"},
{"bin":591,"name":"ENSACAT00000003920","chrom":"scaffold_111","strand":"+","txStart":814576,"txEnd":826972,"cdsStart":814576,"cdsEnd":826972,"exonCount":3,"exonStarts":"814576,825125,826845,","exonEnds":"814594,825247,826972,","score":0,"name2":"ENSACAG00000003945","cdsStartStat":"incmpl","cdsEndStat":"cmpl","exonFrames":"0,0,2,"},
{"bin":591,"name":"ENSACAT00000004042","chrom":"scaffold_111","strand":"-","txStart":849731,"txEnd":881887,"cdsStart":849731,"cdsEnd":881887,"exonCount":24,"exonStarts":"849731,851343,855421,856165,857842,858090,861054,861943,862949,863773,865029,865639,867414,868216,872220,873601,874396,876850,877105,877711,878919,879681,881320,881738,","exonEnds":"849809,851460,855511,856279,857947,858201,861157,862027,863026,863866,865171,865722,867525,868368,872360,873738,874600,876994,877263,877850,878993,879847,881471,881887,","score":0,"name2":"ITGA2","cdsStartStat":"incmpl","cdsEndStat":"incmpl","exonFrames":"0,0,0,0,0,0,2,2,0,0,2,0,0,1,2,0,0,0,1,0,1,0,2,0,"},
{"bin":591,"name":"ENSACAT00000004050","chrom":"scaffold_111","strand":"-","txStart":883724,"txEnd":897808,"cdsStart":883724,"cdsEnd":897808,"exonCount":5,"exonStarts":"883724,885433,889264,889742,897701,","exonEnds":"883858,885548,889356,889852,897808,","score":0,"name2":"ENSACAG00000004086","cdsStartStat":"incmpl","cdsEndStat":"incmpl","exonFrames":"1,0,1,2,0,"}
]);







{kind=link}