In this post, I will present how I wrote a standalone XUL application: it's a simple GUI translating a DNA sequence to a proteic sequence. (Wikipedia:) XUL stands for "XML User Interface Language". It's a language developed by the Mozilla project which operates in Mozilla cross-platform applications, it relies on multiple existing web standards and technologies, including CSS, JavaScript, and DOM. Such reliance makes XUL relatively easy to learn for people with a background in web-programming and design.
A XUL standalone application has not the barreers of security of the web browsers: It can open/save/write/read a file, create a database (via sqlite/mozstorage), etc...
This post was mostly written with the help of the xulrunner tutorial (https://developer.mozilla.org/en/Getting_started_with_XULRunner).
The complete source code of this application is available at: http://code.google.com/p/lindenb/source/browse/#svn/trunk/proj/tinyxul/translate.
File hierarchy
tinyxul/
+translate/
application.ini
+chrome/
chrome.manifest
+translate
translate.xul
+defaults
+preferences/
prefs.js
- application.ini: provides metadata that allows XULRunner to launch the application properly.
- prefs.js: the preferences file
- translate.xul: the xul file containing the layout of the application and, in this case, the javascript methods
translate/application.ini
This file provides metadata that allows XULRunner to launch the application properly.
[App]
Vendor=lindenb
Name=translate
Version=0.1
BuildID=20090209
ID=translation@pierre.lindenbaum.fr
[Gecko]
MinVersion=1.8
translate/defaults/preferences/prefs.js
The preference file contains among other things, the URI of the main xul window to be opened when the application starts
pref("toolkit.defaultChromeURI", "chrome://translate/content/translate.xul");
translate/chrome/translate/translate.xul
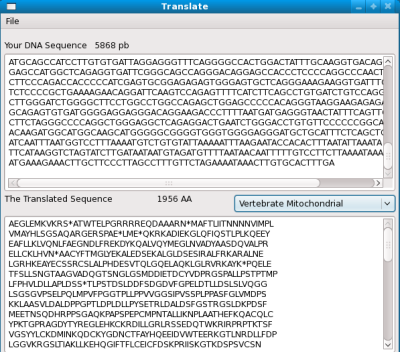
The window is defined with the XML/XUL layout. At the end, it will look like this:
The application is a <window>, it contains a <menubar>, two <textbox> (for the DNA and the proteic sequences) and a <menulist> where the user select the Genetic Code (standard, mitochondrial...)
<?xml version="1.0"?>
<?xml-stylesheet href="chrome://global/skin/" type="text/css"?>
<window id="main" title="Translate" width="800" height="600"
xmlns:html="http://www.w3.org/1999/xhtml"
xmlns="http://www.mozilla.org/keymaster/gatekeeper/there.is.only.xul"
onload="init()"
>
<script>(...)
</script>
<toolbox flex="1">
<menubar id="sample-menubar">
<menu label="File">
<menupopup id="file-popup">
<menuitem label="New" oncommand="doNewWindow();"/>
<menuitem label="Save As..." oncommand="doSaveAs();"/>
<menuseparator/>
<menuitem label="Exit" oncommand="window.close();"/>
</menupopup>
</menu>
</menubar>
</toolbox>
<vbox flex="13">
<hbox><label control="dnaseq" value="Your DNA Sequence" label="Your DNA Sequence" /><label id="dnalength" flex="1" value="0 pb"/></hbox>
<textbox flex="6" id="dnaseq" rows="5" multiline="true" onchange="doTranslate()" oninput="doTranslate()"/>
<hbox>
<label flex="1" control="protseq" value="The Translated Sequence"/>
<label id="protlength" flex="1" value="0 AA"/>
<menulist flex="1" oncommand="currentGeneticCode=GeneticCode[selectedIndex];doTranslate();">
<menupopup id="gcpopup">
</menupopup>
</menulist>
</hbox>
<textbox flex="6" id="protseq" rows="5" multiline="true" readOnly="true"/>
</vbox>
</window>
An array of genetic codes is stored as a javascript array (BTW, thanks to
Brad Chapman and
PJ Davis for their suggestions):
/* via ftp://ftp.ncbi.nih.gov/entrez/misc/data/gc.prt */
var GeneticCode=[
{
name: "Standard" ,
ncbieaa : "FFLLSSSSYY**CC*WLLLLPPPPHHQQRRRRIIIMTTTTNNKKSSRRVVVVAAAADDEEGGGG"
},
{
name: "Vertebrate Mitochondrial" ,
ncbieaa : "FFLLSSSSYY**CCWWLLLLPPPPHHQQRRRRIIMMTTTTNNKKSS**VVVVAAAADDEEGGGG"
},
{
name: "Yeast Mitochondrial" ,
ncbieaa : "FFLLSSSSYY**CCWWTTTTPPPPHHQQRRRRIIMMTTTTNNKKSSRRVVVVAAAADDEEGGGG",
},
{
name: "Bacterial and Plant Plastid" ,
ncbieaa : "FFLLSSSSYY**CC*WLLLLPPPPHHQQRRRRIIIMTTTTNNKKSSRRVVVVAAAADDEEGGGG",
}
];
Each time the DNA sequence is modified, the method
doTranslate is called.
function doTranslate()
{
var ncbieaa= currentGeneticCode.ncbieaa ;
var dna=document.getElementById('dnaseq').value;
var prot="";
var i=0;
var aa="";
var dnalength=0;
var protlength=0;
while(i< dna.length)
{
var c= dna.charAt(i++);
if("\n\t \r".indexOf(c)!=-1)
{
continue;
}
dnalength++;
aa+=c;
if(aa.length==3)
{
prot+=translation(ncbieaa,aa);
protlength++;
if(protlength % 50==0) { prot+="\n";}
aa="";
}
}
document.getElementById('protseq').value = prot;
document.getElementById('protlength').value = protlength+" AA";
document.getElementById('dnalength').value = dnalength+" pb";
}
And because, this is a standalone application, the user can
SAVE the sequence of the protein.
function doSaveAs()
{
try {
const nsIFilePicker = Components.interfaces.nsIFilePicker;
var fp = Components.classes["@mozilla.org/filepicker;1"]
.createInstance(nsIFilePicker);
fp.init(window, "Save As...", nsIFilePicker.modeSave);
var rv = fp.show();
if (!(rv == nsIFilePicker.returnOK || rv == nsIFilePicker.returnReplace) ) return;
var file = fp.file;
var foStream = Components.classes["@mozilla.org/network/file-output-stream;1"].
createInstance(Components.interfaces.nsIFileOutputStream);
foStream.init(file, 0x02 | 0x08 | 0x20, 0666, 0);
var data=document.getElementById('protseq').value;
foStream.write(data, data.length);
foStream.close();
} catch(err){ alert(err.message);}
}
Testing the application
To launch the application, call
xulrunnerxulrunner translate/application.ini
Or firefox with the
app option
firefox -app translate/application.ini
I guess there should have a way to package this application in a jar/zip, but I still haven't found a way to to this.
That's it !
Pierre